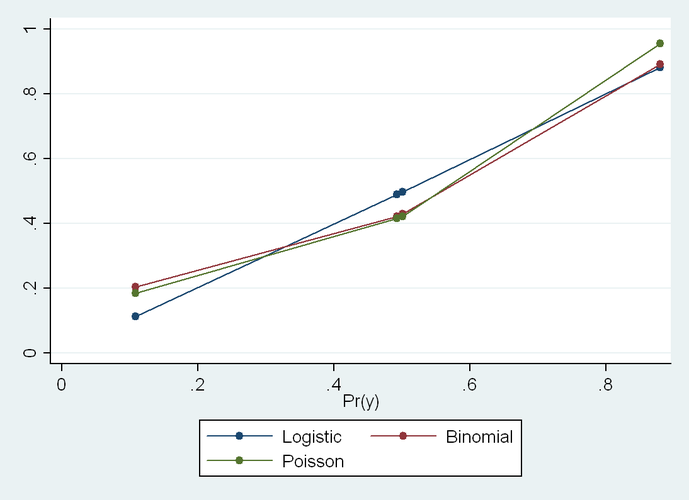
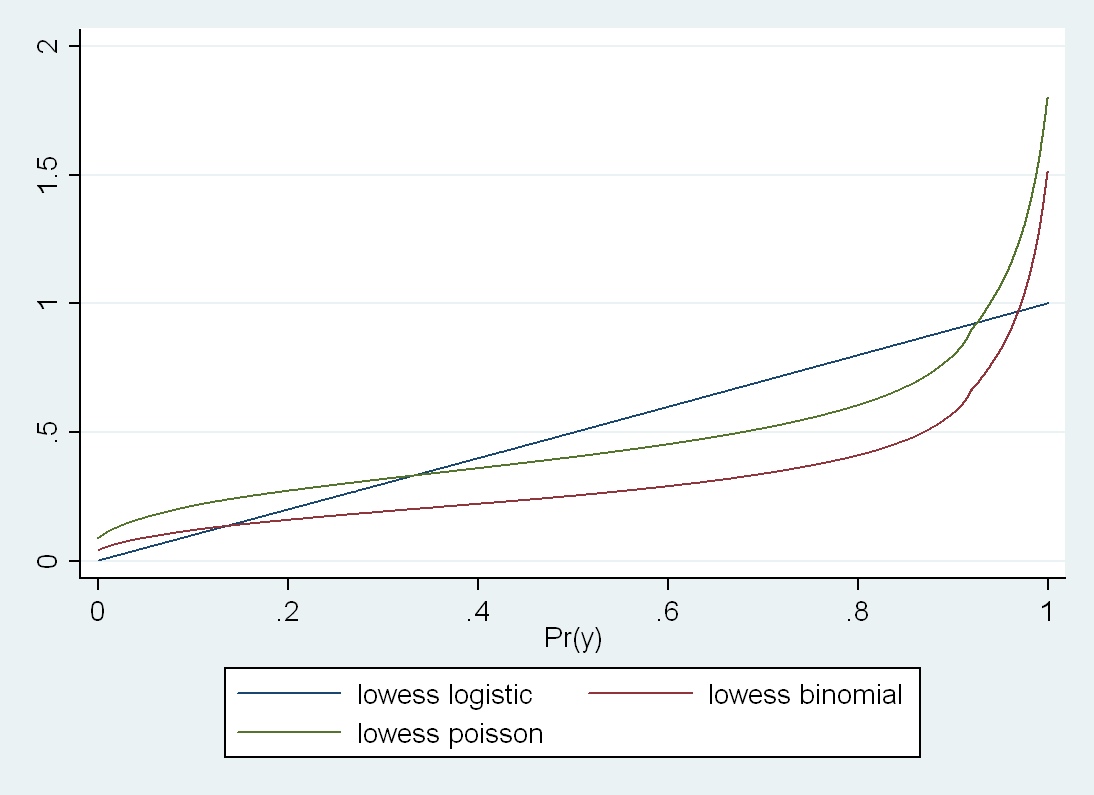
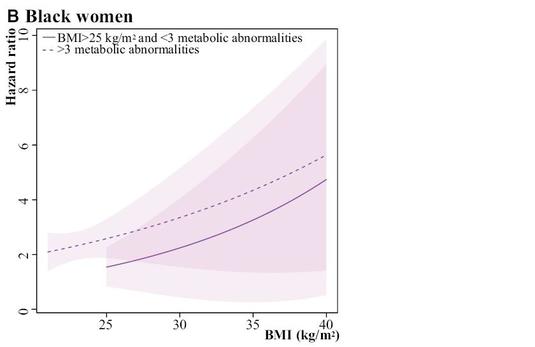
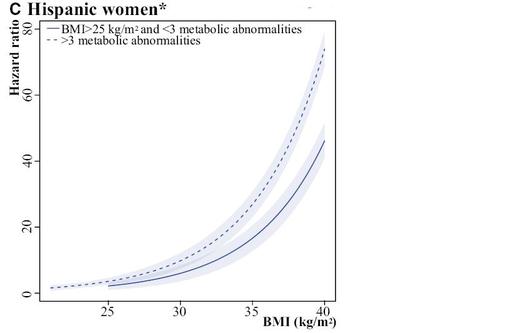
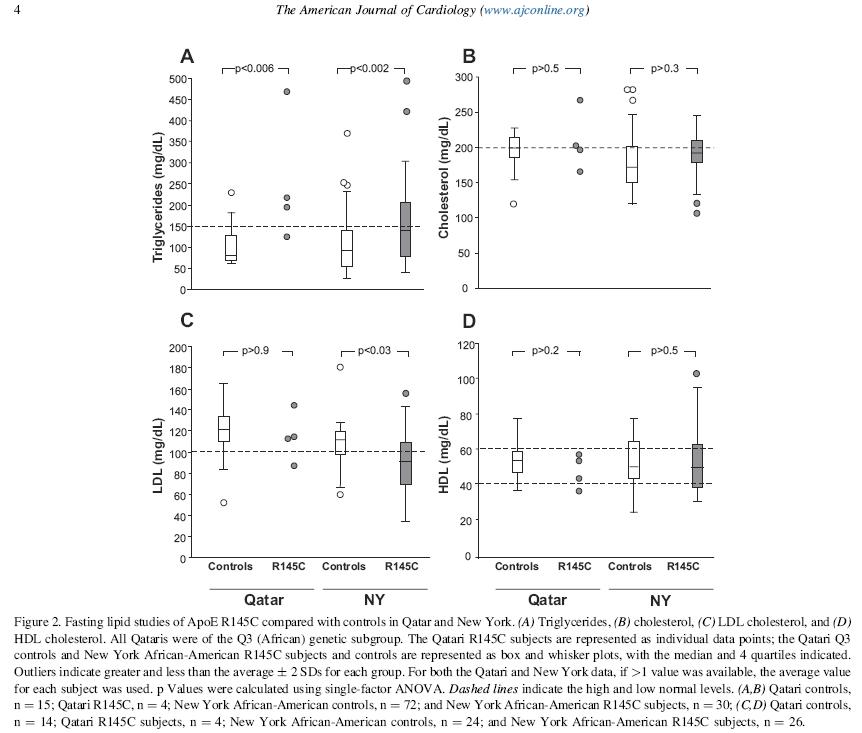
Some of my best friends are fixed-effects Nazis, but I have long been worried about the potential loss of power in situations where there are relatively few observations per cluster and fixed effects drop out all of the concordant strata. You might gain some validity, but at the price of precision, and therefore do worse in terms of MSE. Now I see a new paper that finally addresses this trade-off in greater detail. The Hausman test is treated with great reverence in some quarters, but it is a significance test, after all, and so has all of the usual weaknesses of significance testing, such as the potential for low power to correctly reject the null in some settings, or excessive power to reject a trivially false null in other situations.
Often when faced with an unsatisfactory binary choice, the best solution may be to go a third way. In this context, it could be the so-called "hybrid model", which delivers the validity advantages of the fixed-effects model with the parsimony (and thus precision) advantages of the random effects model. There are ways that this model can go wrong, too, but in many situations, you can have your cake and eat it, too. Presenting this result to a Francophone audience last year, I learned that the way to express that latter sentiment is that you can have both the butter and the butter money.
Often when faced with an unsatisfactory binary choice, the best solution may be to go a third way. In this context, it could be the so-called "hybrid model", which delivers the validity advantages of the fixed-effects model with the parsimony (and thus precision) advantages of the random effects model. There are ways that this model can go wrong, too, but in many situations, you can have your cake and eat it, too. Presenting this result to a Francophone audience last year, I learned that the way to express that latter sentiment is that you can have both the butter and the butter money.


 RSS Feed
RSS Feed